Ab initio structural optimization at finite temperatures based on anharmonic phonon theory: Application to the structural phase transitions of BaTiO$_3$
Abstract
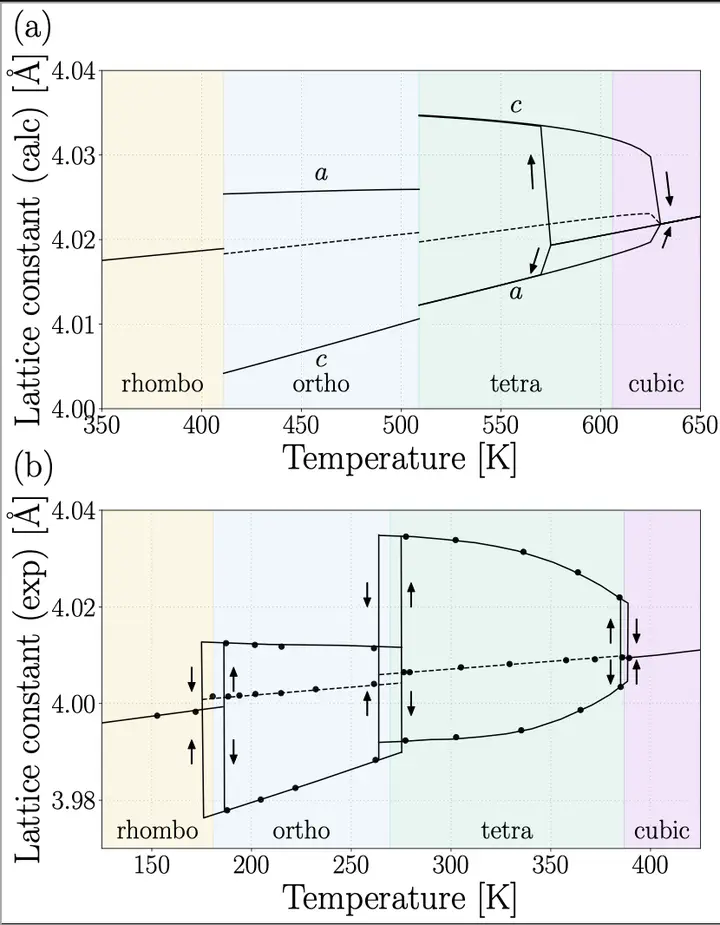
We formulate a first-principle scheme for structural optimization at finite temperature (T) based on the self-consistent phonon (SCP) theory, which accurately takes into account the effect of strong phonon anharmonicity. The T dependence of the shape of the unit cell and internal atomic configuration is determined by minimizing the variational free energy in the SCP theory. At each optimization step, the interatomic force constants in the new structure are calculated without running additional electronic structure calculations, which makes the method dramatically efficient. We demonstrate that the thermal expansion of silicon and the three-step structural phase transitions in BaTiO3 and its pressure-temperature (p-T) phase diagram are successfully reproduced. The present formalism will open the way to the nonempirical prediction of physical properties at finite T of materials having a complex structural phase diagram.
Terumasa Tadano
Researcher of Materials Science
My research interests include development of computational methods and softwares for predicting thermal properties of solids, and application of machine-learning methods to material science study